Could This Radical New Approach to Alzheimer’s Lead to a Breakthrough?
In a small lab in Jackson Hole, Wyo., 65-year-old Paul Cox believes he’s closing in on a treatment that might prevent Alzheimer’s disease. And ALS. And a host of other neurodegenerative diseases, for that matter. Cox, we should point out, isn’t a neurologist. He isn’t a physician of any kind. He doesn’t work at a big drug company or an academic medical center or a government laboratory. His ideas come from so far outside the mainstream of neurological research that you might think he’s crazy or deluded or worse. But then, some very credible people think he might be on to something big—which might make the improbable, quixotic story you are about to read one of the most important as well.
Our unusual tale begins with ethnobotany: the study of the way indigenous people use plants in their customs and diet. You see, Cox is an ethnobotanist, and a darn good one by all accounts. “You’d enjoy walking through a jungle with me,” he once told me. He’s a cheerful gray slouch of a man, quick-witted and sincere, given to club ties and blockish suits when he’s not rocking a fleece. But neurology? When it comes to the study of neurons—the critical cells of the central nervous system that degenerate and die in diseases such as Alzheimer’s and ALS—Cox describes himself as something of a piker. “One colleague says I know about as much neurology as a neurologist’s spouse,” he added with a grin.
Nonetheless, neurons are precisely what you’ll find Cox and a covey of researchers studying at his nonprofit Brain Chemistry Labs. If you happen to be visiting Jackson this winter, you’ll recognize the lab by the cartoonish wood carving of a bespectacled bear (holding a beaker, naturally) just above the front portico. You might even spot a wealthy local patron wearing one of the lab’s “Serine Dipity” sweatshirts. That’s a wordplay on L‑serine, an amino acid that serves critical functions in the central nervous system, among other things. That’s the second strange part of this story: How extraordinarily unlikely and yet wonderful would it be if Cox and his colleagues were right—and the best prevention for some of these terrifying diseases turns out to be a naturally occurring protein building block rather than a high-priced drug?
You can buy a kilo of powdered L-serine for $53 on Amazon. A Serine Dipity sweatshirt, on the other hand, will cost you a $150,000 donation to Cox’s lab. Which leads us to the third twist in this marvelously odd tale. The sweatshirt buyers (and Cox’s wealthy backers) seem to believe just as fervently in the man’s innovative research model as they do in his purported cure. Indeed, it’s fair to say that whether or not Cox’s theory pans out, the style of medical investigation he’s pioneering is gaining fans—even in some traditional and elite academic quarters. So if Cox and his colleagues do push the science forward on Alzheimer’s, ALS, or any other neurological disease even a little, it may have an added benefit of offering the culture of medical research a fresh model to emulate.
And that—in a nutshell—is what the Paul Cox story is all about.
Cox’s interest in neurodegeneration began when he set out to solve a puzzle that had bedeviled researchers for decades: Why did an extraordinary number of the Chamorro people of Guam develop an odd hybrid of ALS and Alzheimer’s symptoms? Cox’s answer: They had been poisoning themselves every time they indulged in their greatest culinary delight, a bat boiled in milk—eyeballs, wings, and all. That was 16 years ago. Since then, Cox has been trying to see if that insight could eventually lead to some kind of treatment against brain diseases.
Working on a tiny budget, Cox has built a consortium of 50 scientists from a wide range of disciplines, who share their unpublished research with one another and push Cox’s theories in directions he never would have anticipated. Within this loose-knit group, the spirit of inquiry seems to thrive, uninhibited by strictures that rein in scientists in academic research centers and pharmaceutical labs. “He’s a visionary,” said Deborah Mash, who runs the Brain Endowment Bank at the University of Miami’s Miller School of Medicine and who has worked with Cox on several experiments. “I was a skeptic. But he’s a fiercely intelligent man. The way he’s pushed this forward is unbelievable.” Cox’s “virtual pharma,” as he calls it, has fostered a more innovative, organic, and patient-focused form of scientific research than what’s often found at the world’s leading drug companies, its members say.
Those companies have failed miserably in their own efforts to attack Alzheimer’s. The FDA has approved just five treatments for Alzheimer’s, and they provide only limited, temporary relief. The agency hasn’t signed off on any new ones since 2003, despite more than 500 clinical trials of Alzheimer’s drugs. In 2018 alone, trials of once-high-profile drugs made by AstraZeneca, Eli Lilly, Johnson & Johnson, Merck, Takeda, and others collapsed or faded away in a statistical whimper. Some big companies, including Pfizer, have completely abandoned the field. (For more on this epic washout, see “Can Biogen Beat the Memory Thief?”)

What do these serial failures have in common? The great majority of the drugs were built on a single idea, the “amyloid hypothesis,” which posits that clumps of protein fragments called beta-amyloid—which are found in the brain of every Alzheimer’s patient—are the primary cause of the disease. (Another hallmark is the presence of neurofibrillary tangles of a protein called tau.) The amyloid theory is based on decades of perfectly good science, and the idea that if you eliminate those plaques you might also slow or reverse the disease still holds sway. But it’s not the only science—and targeting these plaques directly may not ultimately be the best (or only) way to fend off or treat Alzheimer’s.
For decades, though, Big Pharma hasn’t been very interested in less conventional theories. Seeking an enormous payout of perhaps $10 billion a year in sales, they have thrown thousands of scientists and billions of dollars at this one idea, again and again, with no luck.
“You know that definition of insanity?” Cox asked, the first time we met. “Doing the same thing over and over again despite getting the same results? Each trial is a billion bucks; each targets the same thing. None have worked. It seems to me that if you’d put in a billion bucks and failed, you’d say, ‘Let’s try something else.’ ”
If there is any good news about Alzheimer’s, it might be this: After three decades of cureless consensus, the scientific community may finally be ready to seriously consider alternative approaches. One sign of change has been the entreaties in top-tier journals ranging from The New England Journal of Medicine to Brain to Frontiers in Neuroscience to rethink the orthodoxy. (As a New England Journal editorialist put it: “We may very well be nearing the end of the amyloid-hypothesis rope, at which point one or two more failures will cause us to loosen our grip and let go.”) Another sign, perhaps, is the willingness of scores of scientists to sign on to the exploration of a bizarre moonshot of a theory born in the rain forests of Guam.
His mother had died of cancer in 1985, and Coles’s call to arms offered a way forward apart from grieving. So he grabbed some paper and began jotting down his experiences, interests, and talents. “I’m fluent in a couple of Polynesian languages, I’m a marine forest biologist, I’ve studied with the world’s greatest ethnobotanist, and I really want to defeat disease,” he recalled. “If I become an oncologist, maybe I can help dozens of people. If I could discover a new drug, I could help millions of people. What are the chances of that? Oh, about next to zero. But why not give it a shot?”
Two months after his mother’s death, he, his wife, Barbara, and their three kids set off for Falealupo, a tiny village on Savai’i, a Samoan island where they would live, off and on, for several years. The funding came from a 1985 Presidential Young Investigator Award, presented by President Reagan.
Cox didn’t discover a cure for cancer in Samoa. He did, however, find a substance in tree bark that local healers ground into a salve, which Cox suspected might have activity against HIV. (He later licensed the compound to the AIDS Research Alliance of America, but it was never developed into a drug.) He also brokered a deal that helped save 30,000 acres of Samoan rain forest—home to many native species, including Pteropus samoensis, a flying fox, or genus of bat, whose wingspan can stretch nearly three feet wide. Cox and a tribal chief named Fuiono Senio were awarded the Goldman Environmental Prize for the rain forest agreement.

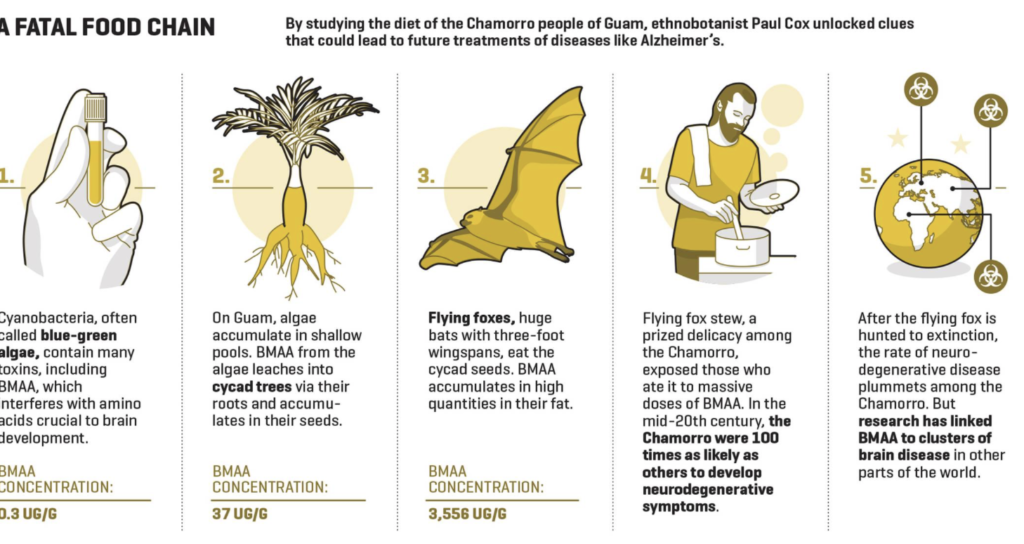
Cox’s interest in bats led him to Guam, and to the mysterious ailment the Chamorro people called lytico-bodig. In the years after World War II, the Chamorro were up to 100 times as likely as people elsewhere in the world to develop symptoms often associated with neurodegenerative diseases like ALS, Alzheimer’s, and Parkinson’s: slurred speech, facial paralysis, loss of motor skills, immobility and dementia. Believing that this cluster might hold essential clues to neurodegeneration, scientists advanced several theories. Some targeted a toxin found in the seeds of local cycad trees. Called BMAA, it killed nerve cells in lab tests and induced symptoms of lytico-bodig when fed to monkeys. The Chamorro cleaned the seeds thoroughly before grinding them into a flour for their version of tortillas. But later research suggested that humans would have to ingest, literally, a ton of cycad flour each month for the toxin to have any effect, and the purported BMAA link fell out of favor.
Cox approached the mystery through the lens of ethnobotany—examining the Chamorro not in the clinic, but in their culture. “And we discover that the flying fox is the most important item in their whole diet,” he said. “They identify themselves as the hunters of flying foxes. One village elder told me, ‘You don’t get this. I would not sell one of those for any price. If I had one, I would lock the door, bolt the windows, cook it, and eat it, and people would be trying to break in to get some.’ ”
Cox believed that this culinary predilection might explain lytico-bodig. One clue was that only older generations of the Chamorro got ill. They had hunted the native bats into extinction. Young Chamorro, who hadn’t grown up feasting on those flying foxes, weren’t getting sick. A second clue was that the Guam bats lived on cycad seeds. If, as Cox believed, BMAA (or another noxious substance) accumulated and magnified over time in bat fat, then every bowl of flying fox stew was toxic. In 2002, he and Oliver Sacks, the late neurologist and author of such books as Awakenings and The Man Who Mistook His Wife for a Hat, published a paper in the journal Neurology that laid out his theory.
Over the next two years, Cox set out to confirm his thesis with Sandra Banack, another bat-loving biologist, and Canadian chemist Susan Murch. In Neurology, they reported finding massive levels of BMAA in museum specimens of the bat. They subsequently discovered BMAA in the brain tissue of Chamorro who had died of lytico-bodig—and also, notably, in the brains of Canadian Alzheimer’s victims. (The toxin, meanwhile, was nowhere to be found in the brains of Chamorro and Canadians who had died of other causes.) The team even made a discovery that seemed to link lytico-bodig to brain diseases around the world. Cycad trees get their sustenance via strange, coral-like, aerial roots. Cox found cyanobacteria, the oldest organism on earth, in those roots.
Cyanobacteria, which are often referred to as blue-green algae, are all around us, in oceans and lakes, in puddles and ponds, even under the crust of deserts from Kuwait to Arizona. And cyanobacteria are loaded with toxins, including BMAA. The Chamorro were just getting ultrahigh doses of a toxin that the rest of us are exposed to all the time. If Cox was right, every green stinky body of water around the world might harbor an insidious source of neurological disease. “It was like staring into the abyss,” he said.
While Cox undertook this initial research, he also had a day job: director of the National Tropical Botanical Garden, a group of five preserves in Hawaii and Florida set aside by congressional mandate for research and conservation. Cox kept his employers abreast of his investigations, and eventually, Doug Kinney, a retired investment banker who chaired the garden’s board, decided that he should move on. “Paul was okay as a garden director,” Kinney told me. “But spending time thinking about who would take care of a particular plot of nasturtiums is not what a great scientific mind ought to be doing.”
“I would not sell one of those for any price,” a village elder said to the Chamorro bat delicacy. “If I had one, I would lock the door, bolt the windows, cook it, and eat it.”
Kinney and a couple of friends, including Bill Egan, the former EVP of Johnson & Johnson’s worldwide consumer products division, told Cox they’d fund a lab where he could research his theory linking the BMAA toxin and neurological disease. They wouldn’t hobble the lab with the red tape typically faced by researchers at pharmaceutical companies and academic labs. Cox and his researchers would decide what experiments to conduct, they’d get new equipment when they asked for it, and neither the board nor Cox would expect any commercial return. The scientist, in turn, promised he’d be efficient; the lab, which was launched in 2006, has an annual budget of around $2.5 million.
Kinney, Egan, and the other initial funders weren’t the only people fascinated by Cox’s tale of the Guam puzzle. Cox is a good storyteller—at Harvard, he twice won the prestigious Bowdoin Prize for essay writing (other winners include Ralph Waldo Emerson and John Updike). And he has attracted a fair amount of publicity, including from Time magazine, which once named him one of 11 “Heroes of Medicine.” Early on, criticism accompanied the attention, often from scientists accusing him of dubious methods and bad science. “Every time [he] comes up with another award or a big glossy story about him, we all just cringe,” one told The New Yorker in 2005. I tried to contact several of his critics for this story, but none returned my emails or phone calls.
Cox, who earned a Ph.D. in biology from Harvard and undergraduate degrees in botany and philosophy from Brigham Young University, acknowledges such skepticism—and seems even to welcome it. Doubt and derision are helpful reminders for scientists—reminders not to be trapped by your own ideas and certainty: “It’s really important, as a scholar and a scientist, to have a contour map of your knowledge,” he told me. “And it’s just as important to have a contour map of your ignorance.”
As he pursued his scientific inquiry on BMAA, he began cobbling together a group of scientists that could fill in the many gaps in his own training. He started with neurologists at the Karolinska Institute in Stockholm. Since then, he told me, “I’ve gone to over 50 people in 28 labs in a dozen countries with the same pitch: ‘Hi, please stop what you’re doing. Help us solve Alzheimer’s and ALS.’”
By all accounts, he’s persuasive. “In 2008, he came to meet us in Sydney,” said Rachael Dunlop, a molecular biologist in Australia. Cox was trying to understand just how the toxin BMAA did its damage in the brain. He believed that it insinuated itself into protein chains in place of one of the 20 standard amino acids, causing misfolding that can trigger the death of neurons. He didn’t know which of the 20 was being displaced, although he suspected glutamate, an important neurotransmitter. Dunlop and her then boss, Ken Rodgers, were expert on this kind of misincorporation, so Cox asked them if they’d investigate. “It’s so gripping when he tells you the story about Guam and Oliver Sacks and the Chamorros and cyanobacteria—how could you not want to work on the project, right?” says Dunlop. “It’s the ultimate scientific detective story. That’s what did it for us.” The research she and Rodgers conducted for Cox proved critical—and also proved him wrong. BMAA was passing for L-serine, not glutamate. Rodgers and Dunlop had handed Cox a potential treatment to combat his toxin. Dunlop eventually went to work for Cox in Jackson, while Rodgers now directs a lab at Sydney’s University of Technology.

Cox is the consortium’s ringleader, emcee, flack, and switchboard operator. He says he’s on email or phone calls with a handful or two of the scientists every week, learning about new research, suggesting new avenues to pursue, and connecting them to others in the group. The consortium gathers once a year, often in Jackson but sometimes in places like Johannesburg or Stockholm. “We’re all in different fields,” marine biologist Larry Brand told me. “We all present our results and try to connect the dots on everything from causes of algae blooms to medical problems to possible prevention and treatment.” Brand’s work has evolved as a result of these collaborations. A decade ago, when he first joined the consortium, Brand was trying to understand what causes the huge algae blooms that Florida sees so often. Now he’s trying to figure out how much BMAA is getting into the food chain via crabs, shrimp, and other marine life that can be found in those blooms. “Paul’s something of a Renaissance man,” Brand told me. “He’s very knowledgeable in a lot of different fields, and he’s very good at connecting the dots.”
Neurologist Aleksandra Stark, who runs the Alzheimer’s clinic at the Dartmouth-Hitchcock Medical Center in Hanover, N.H., attended her first conference last October. “It’s unbelievable,” she said. “All these brilliant people get together and talk about their research around BMAA and cyanobacteria. There was stuff on zebra fish, on cyanobacteria carried by different species of butterflies, on all the various toxins found in blue-green algae. It spanned all domains of science. It was kind of ridiculous—in a good way.”
Cox’s own work has now been cited by other researchers more than 12,000 times in scientific journals. But it’s the consortium as a whole that has really turned his initial insight about the Chamorro into an expansive body of research:
• In Sweden, neuropharmacologist Eva Brittebo revealed that rodents dosed with high levels of BMAA develop neurofibrillary tangles and behavioral aberrations—but only once they become adults, mimicking the long latency period seen in humans who develop Alzheimer’s.
• Dartmouth neurologist Elijah Stommel pinpointed epidemiological clusters of ALS around certain lakes in New England that have had cyanobacteria blooms.
• ALS expert Walter Bradley traveled with Cox to Qatar, where they found swaths of blue-green cyanobacteria laden with BMAA under the desert crust. They believe this might help explain a reported spike in ALS among U.S. veterans of 1991’s Operation Desert Storm. They have since found cyanobacteria under desert crust in Arizona and Utah.
• Algae biologist Larry Brand discovered that certain blue crabs off the coast of Florida that are commonly eaten by humans had levels of BMAA as high as the bats in Guam. “If BMAA were a man-made chemical,” Brand told me, “I don’t think it would ever be allowed to be added to food.”
• Cleveland Clinic neurologist Erik Pioro has plotted 1,000 cases of ALS in the northwest corner of Ohio, near Lake Erie, which is polluted with BMAA and several other neurotoxins.
All this research has inspired other scientists as well. A Norwegian team, for example, has looked at how BMAA affects proteins in zebra fish. In Canada, researchers have shown that BMAA is released from algae blooms as the cyanobacteria die. And in 2016, Chinese scientists showed that rats injected with BMAA developed ALS-like symptoms.
Despite such findings, the consortium’s work is far from accepted science. A 2017 review of the literature on BMAA by scientists at an EPA lab in North Carolina’s Research Triangle Park concluded that “the hypothesis of a causal BMAA neurodegenerative disease relationship is not supported by existing data.”
Undeterred, Cox has steered the focus of the Jackson lab to L-serine, which he believes could significantly delay the onset of Alzheimer’s and the progress of its symptoms. The FDA has previously approved the use of L-serine as a safe dietary supplement, and doctors sometimes prescribe it for chronic fatigue syndrome. The Cox team believes L-serine may play a neuroprotective role.
When I met with Cox recently in New York City, he was quick to share some newly published lab research on the role L-serine plays at the cellular level. We spoke over breakfast at the dreary Times Square hotel he frequents when courting funders or accompanying his wife, Barbara, to Broadway shows. “Here’s what we now think is astonishing about L-serine,” Cox said. “It appears to be neuroprotective against all possible protein misfolding. It basically turns on a system in our brains that looks for unfolded proteins and is quickly poised to act on them.”
For Cox, the most powerful illustration of L-serine’s potential is a 2016 study he and the University of Miami’s Mash oversaw on St. Kitts in the British Virgin Islands. A team at an animal research lab there fed bananas loaded with BMAA, L-serine, or a combination of both to vervet monkeys who have a gene that is thought to increase the risk of Alzheimer’s in humans. (The control group got bananas with rice flour.) Monkeys given BMAA showed both the plaques and tangles common to Alzheimer’s patients. But those given an accompanying dose of L-serine had 80% to 90% fewer tangles in their brain tissue, the study found. The results astounded Mash and Cox, so they repeated the effort with another 140 vervets and got comparable results. Their findings were published in the Proceedings of the Royal Society.
Early in 2017, Cox published the results of a six-month clinical trial of L-serine given at varying doses to ALS patients. The Phase I trial, conducted by independent labs in San Francisco and Phoenix, showed once again that L-serine is safe for humans. One piece of data dangled alluringly from the paper, which was published in a respected ALS journal. The four patients who received the highest doses of L-serine (30 grams per day) saw the progress of their symptoms, as measured on a widely used scale known as ALSFRS-R, slow by 85%. The number of patients, in this case, was too small for the finding to reach statistical significance, but if further clinical trials replicate anything close to that percentage, L-serine would slow the progress of symptoms far more than any existing drug, potentially buying patients years of life. (The average ALS patient dies 2½ years after diagnosis.)
Such “ifs” can be tantalizing and dangerous, particularly if the driving hope behind them masks self-deception or persistent blind spots in the science. In the case of the L-serine conjecture, though, we should at least get a little more evidence, one way or another, next year. That’s when a pair of clinical trials currently underway in Hanover, N.H., are due for completion. Dartmouth’s Elijah Stommel is overseeing a Phase II trial of ALS patients taking 30 grams of L-serine a day, while his colleague Aleksandra Stark supervises a Phase II trial of Alzheimer’s patients receiving the same dosage. Starck is 39 and has been seeing Alzheimer’s patients since her neurology residency at University of North Carolina in 2011. “Ultimately, I am hopeful and optimistic,” she said. “There will be some kind of meaningful slowing of the progression of Alzheimer’s within a decade, even if a cure seems like wishful thinking.”
“This is where we stand,” Cox told me. “We think that chronic exposure to BMAA is a risk factor for ALS and Alzheimer’s. It’s not deterministic. It’s like tobacco and lung cancer: If you smoke, you might not get it, and if you don’t smoke, you still might get it. With L-serine, it’s possible that it could significantly reduce our risk of these diseases. It’s cheap and it’s safe, so it could prove to be the molecule of choice for disease prevention. If the research pans out, we could possibly provide L-serine to all people who are deemed at risk of developing the disease in the future.”
Then he added: “There’s lots of L-serine in bacon. Did I mention that?”
By 2002, when Cox and Sacks first proposed their Guam theory, leading pharmaceutical companies were well into their massive, collective bet on the amyloid hypothesis—a theory that, at least in part, dates back more than a century. In 1906, when Alois Alzheimer examined the brain of a woman who had suffered from dementia and died at 51, he found plaques and neurofibrillary tangles (twisted fibers of protein that may impede a neuron’s normal function). These plaques and tangles are the pathological markers of the disease that came to bear his name. In the mid-’80s, researchers identified amyloid-beta as the misfolded protein in plaques and tau as the misfolded protein in tangles. By the end of that decade, many scientists had settled on the accumulation of amyloid as the primary cause of Alzheimer’s.
Many in the field (and perhaps even most) argue that this remains the case—and that the serial failures of drugs targeting this plaque is simply bad luck. Or perhaps blame is owed to faulty measures in the clinical trials that don’t quite capture the drugs’ beneficial effects. Or perhaps the dosing has been wrong—or the therapy given too late in the game. “The evidence for the amyloid hypothesis has continued to strengthen,” I was told by Daniel Skovronsky, chief scientific officer at Eli Lilly. “There is very strong genetic evidence. And imaging data has made clear that amyloid is there in the brain years before the onset of symptoms. If you have amyloid, you’re at risk of developing Alzheimer’s. If you don’t, you’re not.”
Others, however, see the same data points—hundreds of billions of dollars spent, countless hours of human effort, tens of thousands of patients in ineffectual trials—and see a failure of the drug development process, starting in the academic research institutions. “The problem is the way science is done and funded,” said Zaven Khachaturian, editor-in-chief of trade journal Alzheimer’s & Dementia who formerly directed Alzheimer’s research across the National Institutes of Health, during one of several long phone calls. “It’s populated by people who follow the orthodoxy. To get continuous support, scientists must follow existing orthodoxies. Everybody says they value the individual or the maverick, but nobody will fund them because they say it’s a fishing expedition.” Research has shown that evaluators on panels that award government funding to scientists at research universities regularly give higher scores to conservative proposals than to those trying to break new ground.
Caution is rewarded at the corporate level as well. Pharmaceutical companies trying to move a drug from discovery to approval face a daunting and expensive process. After discovery of a molecule that might have disease-altering potential, pharma companies are required by the FDA to vet their compound with a Phase I clinical trial (to test safety), at least one Phase II trial (to establish potential efficacy), and a massive Phase III clinical trial—often involving thousands of patients tracked over two or more years—to verify its effectiveness and prove its safety for a wide market. The process can take a decade or even two and cost hundreds of millions of dollars or more. The great majority of tested compounds don’t make it through.

You could argue, as many have, that the system works, in that unsafe drugs are unlikely to make it through all these hurdles. However, the time and expense can discourage innovation. Pharmaceutical companies believe it’s safer for them to bet on marginal improvements to an existing therapy than to gamble on an unconventional drug. Repeated failures deter exploration even more: ClinicalTrials.gov, the NIH’s official registry for clinical trials, lists just 215 active studies in Alzheimer’s disease in the U.S., vs. nearly 7,000 directed at cancer, where a variety of treatments have successfully lowered age-adjusted death rates.
There’s been a hefty cost to Big Pharma’s fearful orthodoxy on Alzheimer’s. “Billions of dollars have been spent pushing bad drugs into clinical trials,” said Michael Gold, VP of developmental neurosciences at AbbVie. “There’s the opportunity cost—every dollar that you sink into one program, you can’t sink into something else,” he continued. “Drug discovery programs have been terminated. Expertise has been lost. And some of the biggest companies with the best track records of drug development in neuroscience have left the space.”
By betting so heavily on the amyloid thesis, Big Pharma has slighted other approaches that might hold more promise. There has been much less focus, for example, on the tau protein, even though recent studies suggest that tau is a better indicator than amyloid of when symptoms are going to start seriously affecting patients. Of 19 disease-modifying agents now in Phase III Alzheimer’s trials, 10 target amyloid. Just two focus on tau (though there are additional studies now in Phase II).
In the absence of a cure, the pool of Alzheimer’s patients will soar: while 47 million people worldwide live with Alzheimer’s today, 141 million may have the disease in 2050, according to the Alzheimer’s Association. In the U.S. alone, the financial cost of caring for today’s 5.7 million patients is a staggering annual $277 billion. By mid-century, Americans may spend $1.1 trillion annually on Alzheimer’s, a crippling blow to a reeling health care system.
The ultimate cost, of course, is that we are no closer to curing Alzheimer’s than we were 20 years ago. Alzheimer’s still looms as a kind of living death for so many of us. One of every two people over 85 gets the disease, and since Alzheimer’s patients don’t develop new memories, its onset seems like a premature termination of the experience that is supposed to give meaning to our final years. “If you look at this as a public health issue, in terms of are we solving the problem of reducing the disability of patients, we haven’t made a dent,” said Khachaturian, the Alzheimer’s & Dementiaeditor. Since the beginning of this century, annual deaths from heart disease, stroke, and HIV have gone down. Annual deaths from Alzheimer’s disease have increased by 89%. As I was told several times while reporting this story: “Nobody knows an Alzheimer’s survivor.”
Wherever Paul Cox’s exotic-sounding theories might lead, it’s hard not to see in his grassroots international consortium a research model that’s more flexible, responsive, curious, and humbly collaborative than the siloed, conservative approach of the pharmaceutical industry. It would seem a no-brainer that better collaboration among scientists—across disciplines, companies, and countries—is critical to solving this ancient biological mystery. “We have a lot of exciting facts. But they are isolated, and we haven’t connected the dots,” Khachaturian told me. “A model that brings different perspectives from biology, genetics, pharmacology, psychiatry—even physics and chemistry—that’s the kind of thing that’s needed to solve the big problem, the problem of reducing disability caused by dementia. One doesn’t have to judge whether [Cox’s] idea is good or not. His process is important.”
Read: A Trail of Disappointment for Big Pharma
Neurologist Dale Bredesen, a professor at UCLA’s Geffen School of Medicine and author of The End of Alzheimer’s, agrees. “Paul’s work is exciting,” he told me. “Step 1, he’s found a contributor, BMAA. Step 2 is to figure out how you address the insult, and he’s developed L-serine to do that.” Like Cox, Bredesen believes that the amyloid plaques in the brains of Alzheimer’s patients are symptoms of the disease, rather than the cause.
In fact, the steady, accretive science of the Brain Chemistry Labs consortium has become a fixture of academic journals for so long that, to some, it no longer feels so unusual. As physician and author Andrew Weil put it succinctly: “Cox’s work doesn’t feel so far off the mainstream now.”
Far off or not, the globe-trotting ethnobotanist seems forever to be far away. “I’ve gone to every place where we knew there was an increase in neurological disease,” Cox told me during one of our long, rambling conversations in Jackson. “Then one day I thought, ‘Why don’t we go to places that don’t have any record of Alzheimer’s or ALS at all?’ Where are those places? Well, they must be places where people have intact motor neuron systems, which means they can grow to old age. So we went to the village in Japan which has the oldest people.”
Ogimi is an isolated village of fewer than 4,000 people in the Kunigami district of Okinawa, on the northern side of the island. Ogimi advertises itself as the Village of Longevity; it has the most centenarians per capita, according to the World Health Organization. Scores of researchers and reporters have descended on the hamlet, searching for the secrets of a healthy old age. They’ve fingered any number of factors: years of exercise, an intimate community, a matriarchal society, and a diet rich in tofu and sweet potato.
Cox has now visited Ogimi six times. “These people are mind-blowing,” he said. “I go to interview them, and I say, ‘Tell me about the war.’
‘Which war?’ they say.
‘The World War.’
‘Which World War?’
“These women, they move like ballerinas. A 98-year-old who can bend over and touch the mat with her palms. I met a 54-year-old who came to the village from mainland Japan when she married a matriarch’s son. She looks like she’s 19. On a hunch, I ask if she has a sister. She says yes, and when she brings out the photograph, it’s like looking at the portrait of Dorian Gray!” Cox clapped his hands together.
“I got dead serious about looking at their diet,” he said. Cox interviewed dozens of locals, most often over breakfast, lunch, or dinner. He went to the market and bought samples of all the local produce. He even walked the beach to collect seaweed after observing locals doing that at sunset. He shipped it all back to the lab, where his colleagues analyzed the molecular makeup of the Ogimi diet.
“Wouldn’t you know it! The Ogimi people are getting three to four times the level of L-serine that Americans get in their average daily diet,” Cox said. “They have the highest L-serine content of any population that I’ve ever measured. They look unbelievable. And they live forever!”
A version of this article appears in the February 2019 issue of Fortune with the headline “Paul Cox Has A Radical Theory For What’s Causing Alzheimer’s .”